Metagenome Sequencing
Metagenomics is the study of the genomes of a whole microbial community, which has several advantages over single bacterial genomic studies. Microorganisms generally live in symbiotic relationships, and thus metagenomics studies focuses on the genome sequencing of symbiotic microbes instead of a single microbial species. Thus, it does not require the isolation or lab cultivation of individual microbial species.
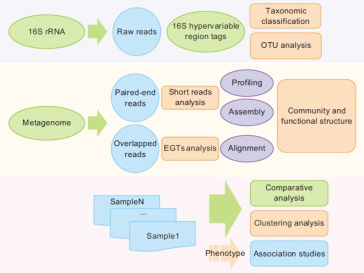
Currently there are two main sequencing strategies used in metagenomics: 16S rRNA sequence analysis using Sanger sequencing technology and metagenomics or metatranscriptomics analysis using NGS technology. The former method allows a general taxonomic classification, but does not detect trace species in a microbial community; therefore, it cannot fully characterize the diversity of gene function within the samples. The latter method allows additional research at the nucleotide level, such as species clustering analysis, gene function analysis, and association studies.
Workflow
Bioinformatics analysis primarily includes: species composition analysis, short reads assembly, gene prediction, gene functional annotation, and statistical analysis of the results. For multiple samples, the comparative analysis between the different samples and the association studies can also be provided.
Bioinformatics
Data analysis includes:
16S rRNA analysis:
We overlap the high quality paired-end reads to form tags from the V3/V6 hypervariable region of the 16S rRNA gene sequence. Taxonomic classification is then performed by aligning these tags to the database and followed by operational taxonomic unit (OTU) analysis based on clustering distance.
Metagenome analysis:
(a) Short-reads analysis: This is based on short-read alignment to the sequenced bacterial genome database where species quantitative analysis is performed first, followed by assembly of the metagenomics reads into contigs and gene prediction from these contigs. Finally, functional annotation and species diversity of the genes are obtained by alignment to data in several databases, including Nt, Nr, KEGG, COG, Swissprot, eggNOG, CAZy, etc.
(b) EGTs analysis: High quality paired-end reads are overlapped to form Environmental Gene Tags (EGTs), after which phylogenetic and functional annotation are performed by aligning to data in several, e.g. Nt, Nr database, etc.