

Exome Sequencing
Cases
Technical Information
Contact Us / Wish List
 BGI is a world-leading provider of Human Whole Exome Sequencing, having sequenced more than 45,000 human exomes to date. Our expertise in human exome sequencing comes from our 15 years of experience doing more whole exome and whole genome sequencing than any institution in the world. Our data analysis prowess is derived from our long history and extensive work in creating innovative and highly effective proprietary approaches and algorithms for analyzing data for genome analysis. We have unparalleled experience and expertise in both the process of large scale NGS sequencing and in analyzing the data generated.
BGI is a world-leading provider of Human Whole Exome Sequencing, having sequenced more than 45,000 human exomes to date. Our expertise in human exome sequencing comes from our 15 years of experience doing more whole exome and whole genome sequencing than any institution in the world. Our data analysis prowess is derived from our long history and extensive work in creating innovative and highly effective proprietary approaches and algorithms for analyzing data for genome analysis. We have unparalleled experience and expertise in both the process of large scale NGS sequencing and in analyzing the data generated.
In addition to Human Exome Sequencing, we provide mouse exome sequencing service using Agilent Mouse All Exon kit and developed a platform for sequencing the exome of monkeys based on our participation in the Chinese rhesus macaque and Cynomolgus macaque genome projects.
While exomes account for 1% of the human genome, protein coding regions contain about 85% of the pathogenic mutations. By selectively targeting DNA sequences that encode proteins, exome sequencing allows for the identification of novel gene mutations associated with both Mendelian disorders and common diseases.
Advantages
- Multiple platforms/technologies are utilized to deliver optimal results:
- Complete Genomics (New!)
- Ion Proton
- Illumina
- Complete Genomics (New!)
- Cost-Effective: Our proficiency and efficiency in exome sequencing allows us to offer our customers comprehensive analysis of whole exons at lower prices than other providers. Researchers can now do more science and get more answers with limited research budgets.
- Industry-leading throughput/ turnaround: Our highest throughput sequencing capacity and rapid project turnaround times enable our customers to advance their research more rapidly and effectively allowing them to attain their goals in a more timely manner
- Strong bioinformatics analysis:We deliver information, not just results. Our advanced bioinformatics team provides accurate standard and advanced analysis with SNP detection over 99% for human research fields.
Platform Options
Having multiple sequencing platforms enables BGI to choose (in consultation with the customer) the most appropriate system to solve their scientific challenges.
The Complete Genomics platform provides industry leading accuracy and comprehensive variation detection ideal for the research that needs:
- Precise mapping of genetic recombination sites
- Accurate identification of de novo disease causing mutations
- Sensitive detection of somatic variants in complex cancer genomes
We currently offer accurate exome sequencing at 100X on the Complete Genomics platform. Free SNP validation of 100+ loci is included to ensure the accuracy of your results, and we will redo sequencing for you at no additional cost if the percentage of correct SNP callings among validated sites falls below 95%.
The Ion Proton platform has rapid sequencing speeds (only 2-4 hours for every sequencing run), and requires only 50-100 ng input DNA. Therefore, it is suitable for projects with a need for:
- Extremely rapid turn-around time
- Analysis of very low amount of DNA
Please check out our hot rapid exome sequencing offer at 100X on the Ion Proton platform, taking you from sample to results in as little as 7 days.
Illumina platform provides high throughput analysis and ample bioinformatics tools are available for a wide range of research analyses. Thus, this platform is suitable for projects with a need for
- Large scale throughput
- Custom bioinformatics analysis tailored for specific research needs
Customer Testimonial
"Knome has been working with BGI for well over three years. Specifically, BGI has been resequencing whole human genomes and exomes for us and it is work they perform exceedingly well. Partnering with BGI has been a positive experience for us and we highly recommend them."
-Ari Kiirikki, VP of Knome
BGI has successfully completed numerous exome sequencing projects, including a Danish study of 1000 patient samples and 1000 controls with the aim of finding rare SNPs associated with metabolic disorders such as obesity and hypertension.
Frequent Mutations of Genes Encoding Ubiquitin-mediated Proteolysis Pathway Components in Clear Cell Renal Cell Carcinoma. Nature Genetics. 44:17-9 (2012).
 The research sequenced whole exomes of ten clear cell renal cell carcinomas (ccRCCs) and performed a screen of ~1,100 genes in 88 additional ccRCCs. Frequent mutations were detected in the ubiquitin-mediated proteolysis pathway (UMPP). The findings highlight the potential contribution of UMPP to ccRCC tumorigenesis through the activation of the hypoxia regulatory network.
The research sequenced whole exomes of ten clear cell renal cell carcinomas (ccRCCs) and performed a screen of ~1,100 genes in 88 additional ccRCCs. Frequent mutations were detected in the ubiquitin-mediated proteolysis pathway (UMPP). The findings highlight the potential contribution of UMPP to ccRCC tumorigenesis through the activation of the hypoxia regulatory network.
Â
Exome Sequencing Identifies NMNAT1 Mutations as a Cause of Leber Congenital Amaurosis. Nature Genetics. 44:972-4 (2012).
 The exome of an individual with Leber congenital amaurosis(LCA) was sequenced and identified nonsense (c.507G>A, p.Trp169*) and missense (c.769G>A, p.Glu257Lys) mutations in NMNAT1,which encodes an enzyme in the nicotinamide adenine dinucleotide (NAD) biosynthesis pathway.It is implicated in protection against axonal degeneration. We also found NMNAT1 mutations inten other individuals with LCA, all of whom carry the p.Glu257Lys variant.
The exome of an individual with Leber congenital amaurosis(LCA) was sequenced and identified nonsense (c.507G>A, p.Trp169*) and missense (c.769G>A, p.Glu257Lys) mutations in NMNAT1,which encodes an enzyme in the nicotinamide adenine dinucleotide (NAD) biosynthesis pathway.It is implicated in protection against axonal degeneration. We also found NMNAT1 mutations inten other individuals with LCA, all of whom carry the p.Glu257Lys variant.
Â
Single-Cell Exome Sequencing Reveals Single-Nucleotide Mutation Characteristics of a Kidney Tumor. Cell. 148:886-95 (2012).
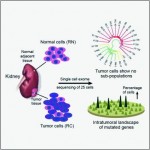 To better understand the intratumoral genetics underlying mutations of ccRCC, single-cell exome sequencing was carried out on a clear cell renal cell carcinoma (ccRCC) and its adjacent kidney tissue. The pilot study demonstrates that ccRCC may be more genetically complex than previously thought and provides information that can lead to new ways to investigate individual tumors, with the goal of developing more effective cellular targeted therapies.
To better understand the intratumoral genetics underlying mutations of ccRCC, single-cell exome sequencing was carried out on a clear cell renal cell carcinoma (ccRCC) and its adjacent kidney tissue. The pilot study demonstrates that ccRCC may be more genetically complex than previously thought and provides information that can lead to new ways to investigate individual tumors, with the goal of developing more effective cellular targeted therapies.
An Integrated Map of Genetic Variation from 1,092 Human Genomes. Nature. 491:56-65 (2012).
 By characterizing the geographic and functional spectrum of human genetic variation, the 1000Genomes Project aims to build a repository that can help understanding the genetic contribution to disease. Up to 98% of accessible single nucleotide polymorphisms at a frequency of 1% in related populations were captured, enabling analysis of common and low-frequency variants in individuals from diverse, including admixed, populations.
By characterizing the geographic and functional spectrum of human genetic variation, the 1000Genomes Project aims to build a repository that can help understanding the genetic contribution to disease. Up to 98% of accessible single nucleotide polymorphisms at a frequency of 1% in related populations were captured, enabling analysis of common and low-frequency variants in individuals from diverse, including admixed, populations.
Â
Sequencing of 50 Human Exomes Reveals Adaptation to High Altitude.Science. 329, 75 (2010).

50 exomes of ethnic Tibetans were sequenced for 18X per individual. Genes showing population-specific allele frequency changes, which represent strong candidates for altitude adaptation, were identified. One single-nucleotide polymorphism (SNP) at EPAS1 shows a 78% frequency difference between Tibetan and Han samples, representing the fastest allele frequency change observed at any human gene to date. This research can help us to prevent and cure the disease of plateau anoxia.
Ion Proton Platform
Illumina Platform
Complete Genomics Platform (For Human Research)
BGI has expanded the application of the Complete Genomics platform, making it possible to perform exome sequencing using this technology. The advantages that the Complete Genomics platform brings to whole-genome sequencing can now be applied to exome sequencing. In addition to the two primary components of this proprietary sequencing technology (i.e., DNA nanoball and combinatorial probe anchor ligation), we integrated exome probe capture with a proprietary library construction process to insert two adaptors into each captured DNA fragment (Figure 1), which enhances the speed of this technology.
Dozens of reports of studies using the Complete Genomics platform have been published in top-tier journals such as Nature, Science, and Cell. More than 15,000 genomes have been sequenced using this platform, producing data that are widely recognized as being highly accurate.

Figure 1. Exome capture and library construction in whole exome sequencing on the Complete Genomics platform
Analysis Workflow
1. Alignment of reads to the reference database with Teramap
2. Local de novo variant assembly of unmatched reads to identify key differences in exons
3. Annotation of all types of variants using entries from a variety of public databases
Advantages
- Industry-leading read accuracy (1 error in 100,000 bases) enabled by the proprietary library construction process in conjunction with the combinatorial probe-anchor ligation technology
- Sensitive detection of allele heterozygosity with proprietary local de novo variant mapping and assembly software
- More accurate and detailed annotation of variants with advanced data management software
- Lower error rate in SNP calling and lower false negative-rates than other platforms
Sample requirements
- DNA amount: ≥3 μg
- Concentration: ≥37.5ng/μL
Recommended sequencing depth
150X
Turnaround time
2 months
Bioinformatics

Cancer Advanced Analysis
- Normal-tumor sample SNV calling, annotation, and statistics
- Normal-tumor sample InDel calling, annotation, and statistics
- Somatic lesser allele fractions (LAF) estimations
In addition, BGI’s innovative bioinformatics experts will be enhancing the data analysis capabilities of the Complete Genomics platform, creating powerful tools to increase the amount of the information obtained from the system.
Â
Ion Proton Platform
Advantages
- Rapid sequencing speed: Only 2–4 hours required for each sequencing run (much faster than the Illumina platform), leading to a significantly shorter turnaround time
- High reproducibility: High correlation of target base coverage depth between technical replicates with correlation coefficient greater than 0.93
- Flexible throughput: The Ion PI™ Chip provides 40–80 million reads, enabling sequencing of 1–2 human exomes per run, without the need to wait for large amounts of samples
- Low required DNA input: Only 50-100 ng DNA for one round of library construction and sequencing
- Advanced bioinformatics analysis: More insightful variant annotations using 10+ public and private databases (e.g., 5000 Exomes and DrugBank)
Sample requirements
- DNA amount: 50-100 ng.
- DNA concentration: ≥1.67 ng/μL
Sequencing strategy
- Sequencing reads: SE 200
- Sequencing coverage: 100X.
Turnaround time
1 week for up to 40 samples
Bioinformatics
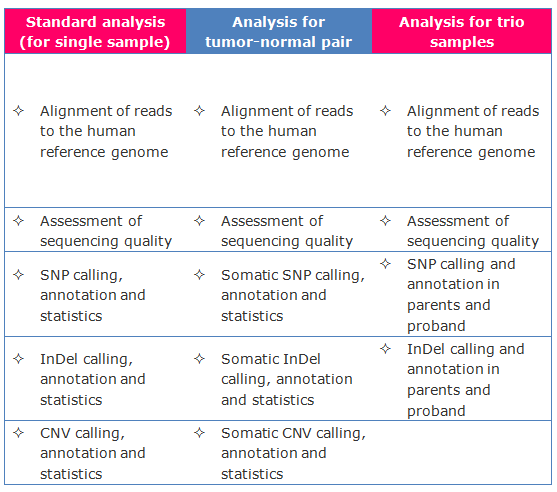
Â
The variant annotation databases are described in the following table.
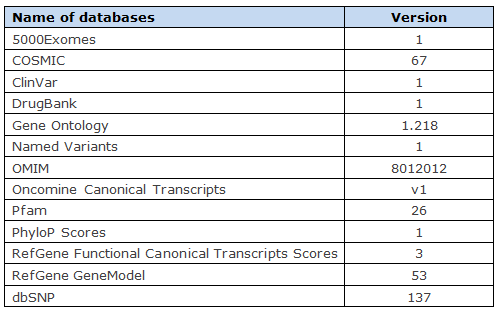
Â
Illumina Platform
Advantages
- High quality sequencing data (Q20>90%,Q30>85%)
- More than 98% of exons covered
- Long sequencing reads (up to PE 150)
- Ample bioinformatics tools available for downstream analysis to meet all research needs
- Low DNA input requirement (starting at 200ng)
Sample requirements
- DNA amount: ≥2.5 µg (standard, although as low as 200 ng is acceptable in some circumstances).
- DNA concentration: ≥37.5 ng/μL.
Sequencing strategy
- Sequencing reads: PE 91/101.
- Sequencing coverage: ≥50X; at least 100X for cancer samples.
Turnaround time
40 days.
Bioinformatics
Standard analysis
- Data filtering (removing adaptors, contamination, and low-quality reads from raw reads).
- Align reads to the human reference genome (UCSC build HG19) using BWA software.
- Assessment of sequencing quality.
- SNP and InDel calling.
- SNP and InDel annotation (annotate each SNP to the corresponding gene functional units in the RefGene database, including nucleotide and amino acid changes).
- SNP and InDel validation and comparison with dbSNP database, 1000 Genomes Project database, publicly available exome databases (ESP), and YH.
- SNP function prediction and sequence conservation analysis.
- Statistical analysis of SNPs and InDels in each functional unit.
Advanced Analysis
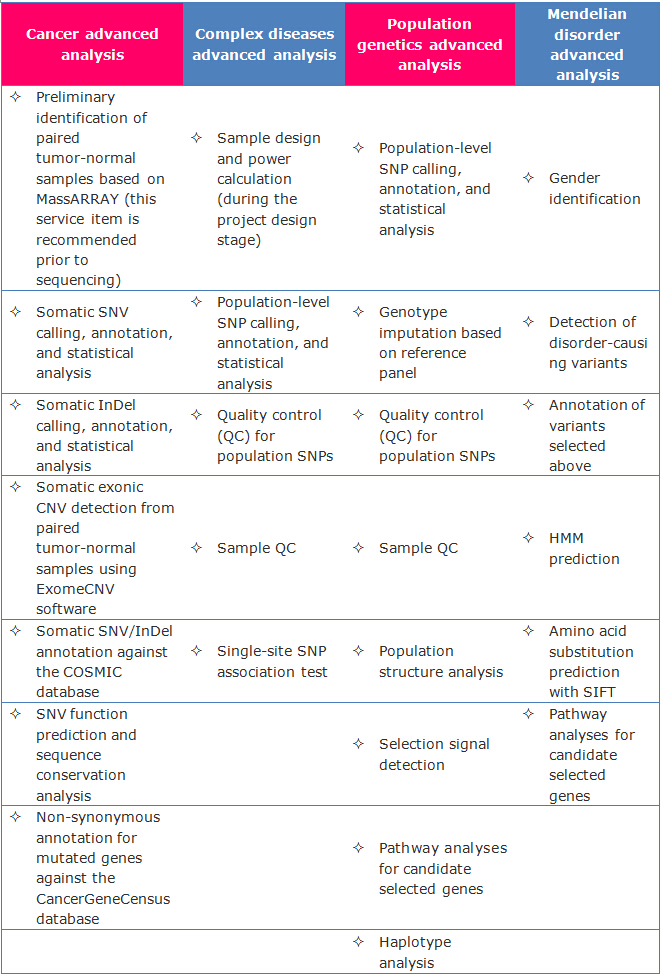
Customized analysis
- De novo mutation analysis based on family samples.
- Customized analysis for complex diseases (applied to large-scale sample design, ≥200 cases & controls are recommended).