

Targeted Proteomics
Cases
Technical Information
Contact Us / Wish List
 Targeted proteomics relies on the selectivity of multiple reaction monitoring (MRM) for recognition of specific analytes and relative or absolute quantification of target proteins. This highly selective and sensitive technology is ideal for verifying biomarkers, discovering low abundance proteins and post-translational modifications, and distinguishing among highly homologous protein isoforms.
Targeted proteomics relies on the selectivity of multiple reaction monitoring (MRM) for recognition of specific analytes and relative or absolute quantification of target proteins. This highly selective and sensitive technology is ideal for verifying biomarkers, discovering low abundance proteins and post-translational modifications, and distinguishing among highly homologous protein isoforms.
Benefits:
- High specificity: Capable of distinguishing among highly homologous protein isoforms.
- High throughput:Dozens of target proteins can be measured during a single analysis.
- High reproducibility: Highly reproducible within and across laboratories and instruments.
- Significance: Targeted proteomics was chosen as the Nature Method of the Year for 2012.
Stress Responsive Proteins Are Actively Regulated during Rice (Oryza sativa) Embryogenesis as Indicated by Quantitative Proteomics Analysis. Plos one. DOI: 10.1371/journal.pone.0074229 (2013).
 Embryogenesis is the initial step in a plant's life, and the molecular changes that occur during embryonic development are largely unknown. To explore the relevant molecular events, we used the isobaric tags for relative and absolute quantification (iTRAQ) coupled with the shotgun proteomics technique (iTRAQ/Shotgun) to study the proteomic changes of rice embryos during embryogenesis. For the first time, a total of 2165 unique proteins were identified in rice embryos, and the abundances of 867 proteins were actively changed based on the statistical evaluation of the quantitative MS/MS signals. The quantitative data were then confirmed using multiple reactions monitoring (MRM) and were also supported by our previous study based on two-dimensional gel electrophoresis (2 DE). Using the proteome at 6 days after pollination (DAP) as a reference, cluster analysis of these differential proteins throughout rice embryogenesis revealed that 25% were up-regulated and 75% down-regulated. Gene Ontology (GO) analysis implicated that most of the up-regulated proteins were functionally categorized as stress responsive, mainly including heat shock-, lipid transfer-, and reactive oxygen species-related proteins. The stress-responsive proteins were thus postulated to play an important role during seed maturation.
Embryogenesis is the initial step in a plant's life, and the molecular changes that occur during embryonic development are largely unknown. To explore the relevant molecular events, we used the isobaric tags for relative and absolute quantification (iTRAQ) coupled with the shotgun proteomics technique (iTRAQ/Shotgun) to study the proteomic changes of rice embryos during embryogenesis. For the first time, a total of 2165 unique proteins were identified in rice embryos, and the abundances of 867 proteins were actively changed based on the statistical evaluation of the quantitative MS/MS signals. The quantitative data were then confirmed using multiple reactions monitoring (MRM) and were also supported by our previous study based on two-dimensional gel electrophoresis (2 DE). Using the proteome at 6 days after pollination (DAP) as a reference, cluster analysis of these differential proteins throughout rice embryogenesis revealed that 25% were up-regulated and 75% down-regulated. Gene Ontology (GO) analysis implicated that most of the up-regulated proteins were functionally categorized as stress responsive, mainly including heat shock-, lipid transfer-, and reactive oxygen species-related proteins. The stress-responsive proteins were thus postulated to play an important role during seed maturation.
Quantitative Analysis of the Human AKR Family Members in Cancer Cell Lines Using the mTRAQ/MRM Approach. J. Proteome Res. 12 (5), 2022–2033 (2013).
Members of human aldo-keto reductase (AKR) superfamily have been reported to be involved in cancer progression, whereas the final conclusion is not generally accepted. Herein, we propose a quantitative method to measure human AKR proteins in cells using mTRAQ-based multiple reaction monitoring (MRM). AKR peptides with multiple transitions were carefully selected upon tryptic digestion of the recombinant AKR proteins, while AKR proteins were identified by SDS-PAGE fractionation coupled with LC-MS/MS. Utilizing mTRAQ triplex labeling to produce the derivative peptides, calibration curves were generated using the mixed lysate as background, and no significantly different quantification of AKRs was elicited from the two sets of calibration curves under the mixed and single lysate as background. We employed this approach to quantitatively determine 6 AKR proteins, AKR1A1, AKR1B1, AKR1B10, AKR1C1/C2, AKR1C3, and AKR1C4 in 7 different cancer cell lines, and for the first time to obtain the absolute quantities of all the AKR proteins in each cell. The cluster plot revealed that AKR1A and AKR1B were widely distributed in most cancer cells with relatively stable abundances, whereas AKR1Cs were unevenly detected among these cells with diverse dynamic abundances. The AKR quantitative distribution in different cancer cells, therefore, may assist further exploration toward how the AKR proteins are involved in tumorigenesis.
Workflow:
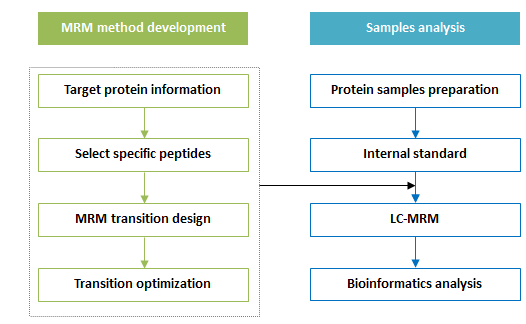
Figure 1. Workflow of Targeted Proteomics
Bioinformatics:
Standard Bioinformatics analysis
- Statistics analysis and evaluation of data
- MRM transition list design
- Target protein identification and quantification
Custom Bioinformatics Analysis
- We can also perform customized analyses to meet specific needs of your projects.
Sample Requirements:
|
Sample Type |
Amount of Sample required |
Comment(s) |
|
Fresh animal tissue (wet weight) |
≥ 100 mg |
For tissue with high impurity or low amount of proteins such as plant root or phloem, 5g sample is needed. |
|
Fresh plant tissue or fungus (wet weight) |
≥ 1 g |
|
|
Microorganism (e.g. bacteria) |
≥ 200 mg |
|
|
Cell |
≥ 3-5×106 cells |
For phosphorylated protein identification, the cell number should be more than 107. |
|
Blood |
Serum / plasma ≥ 500 µl Whole blood ≥ 5 ml |
We do not recommend sending whole blood sample because blood cells can be broken during transport process. |
|
Please remove high-abundance protein before sending extracted proteins and attach gel images before and after the removal of high-abundance protein. |
||
|
Body fluid (e.g. saliva and cerebrospinal fluid) |
≥ 5 ml |
/ |
|
Urine |
≥ 50 ml |
Centrifuge at 1000g for 5min to discard the precipitate before sending your sample to BGI Tech. |
|
Protein extract |
≥ 500 µg (concentration ≥1mg/ml) |
/ |