

Single-Cell DNA Sequencing
Cases
Technical Information
Contact Us / Wish List
Single cell sequencing can facilitate the elucidation of cell lineage relationships. The major applications of this technique include profiling scarce clinical samples (i.e. circulating tumor cells), pre-implantation genetic diagnosis, embryonic development research, and tumor progression analysis.
Advancing the possibilities of single cell sequencing in human disease research, we have developed an innovative end-to-end solution for genomics analysis at the single cell level. Within this solution, multiple displacement amplification (MDA) has been further enhanced and incorporated into BGI’s whole genome amplification (WGA) protocol, which enables uniform amplification of genomic DNA from single cells (from as little as one cell) with negligible sequence bias and maximized genome coverage.
BGI has sequenced hundreds of cells, and the results from applying the new single-cell sequencing method to identify the genetic characteristics of essential thrombocythemia and clear cell renal cell carcinoma have been published in the journal Cell.
Benefits:
-
Less quantity of input DNA required
-
Longer length of amplified product (>10 kb), which is better for CNV and SV detection
-
High accuracy of amplification (error rate is 10-6-10-7)
-
High coverage: >95% bases can be targeted
-
Strict quality control: a QC step is measured by detecting housekeeping genes of WGA products to ensure comprehensive coverage and unprecedented low bias
Applications:
-
Variants calling at the single cell level
-
Pre-implantation genetic diagnosis (PGD)
-
Measuring intra-tumor heterogeneity and guiding chemotherapy
-
Clone evolution analysis during tumor progression
Comparison of Two WGA Methods:
Our WGA method is based on MDA technology, which shows better coverage at low sequencing depth than Multiple Annealing and Looping Based Amplification Cycles (MALBAC) technology.
|
MALBAC |
Next-generation MDA (employed at BGI) |
|
|
Amplified DNA length |
0.5-1.5kb |
2-100kb |
|
Induced artificial sequence |
70bp (35bp on each end of primer sets), thus ~7% of data is useless considering average size of amplified DNA fragments is about 1kb |
None |
|
Whole genome coverage (sequencing depth >25X) |
84%~93% |
~99% |
|
False Positive rate |
4X10-5 |
1X10-5 |
Data Performance:
Data performance of our WGA technology is shown below.Table. Single-Cell Sequencing Data Evaluation on human chromosome 12
|
Sample |
Mean depth |
% of bases at ≥1X |
% of bases at ≥5X |
% of bases at ≥15X |
ADO |
|
Single-cell 1 |
31.56 |
99.74 |
98.26 |
80.39 |
0.43% |
|
Single-cell 2 |
38.42 |
99.5 |
97.44 |
83.92 |
5.21% |
|
Genomic DNA from human tissue (control) |
37.76 |
99.74 |
99.54 |
98.49 |
Discovery of Biclonal Origin and a Novel Oncogene SLC12A5 in Colon Cancer by Single-cell Sequencing. Cell Research. 24(6):701-12 (2014).
Single-cell sequencing is a powerful tool for delineating clonal relationship and identifying key driver genes for personalized cancer management. Here we performed single-cell sequencing analysis of a case of colon cancer. Population genetics analyses identified two independent clones in tumor cell population. The major tumor clone harbored APC and TP53mutations as early oncogenic events, whereas the minor clone contained preponderant CDC27 and PABPC1 mutations. The absence of APC and TP53 mutations in the minor clone supports that these two clones were derived from two cellular origins. Next, we identified a mutated gene SLC12A5 that showed a high frequency of mutation at the single-cell level but exhibited low prevalence at the population level. Functional characterization of mutant SLC12A5 revealed its potential oncogenic effect in colon cancer. Our study provides the first exome-wide evidence at single-cell level supporting that colon cancer could be of a biclonal origin, and suggests that low-prevalence mutations in a cohort may also play important protumorigenic roles at the individual level.
Figure 1. The major tumor clone (red) was characterized by prevailing TP53 and APC mutations, whereas the minor clone (blue) contained mutations in two cancer-related genes, CDC27 and PABPC1. Isolated normal cells (green) did not harbor any detectable mutation.
Single-Cell Exome Sequencing and Monoclonal Evolution of a JAK2-Negative Myeloproliferative Neoplasm.Cell. 873-885 (2012).
Tumor heterogeneity presents a challenge for inferring clonal evolution and driver gene identification. Here, we describe a method for analyzing the cancer genome at a single-cell nucleotide level. To perform our analyses, we first devised and validated a high-throughput whole-genome single-cell sequencing method using single cells from two lymphoblastoid cell lines. We then carried out whole-exome single-cell sequencing of 90 cells from a JAK2-negative myeloproliferative neoplasm patient. The sequencing data from 58 cells passed our quality control criteria, and these data indicated that this neoplasm represented a monoclonal evolution. We further identified essential thrombocythemia (ET) related candidate mutations such as SESN2 andNTRK1, which may be involved in neoplasm progression. This pilot study allowed the initial characterization of the disease-related genetic architecture at the single-cell nucleotide level. Further, we established a single-cell sequencing method that opens the way for detailed analyses of a variety of tumor types, including those with high genetic variations between patients.
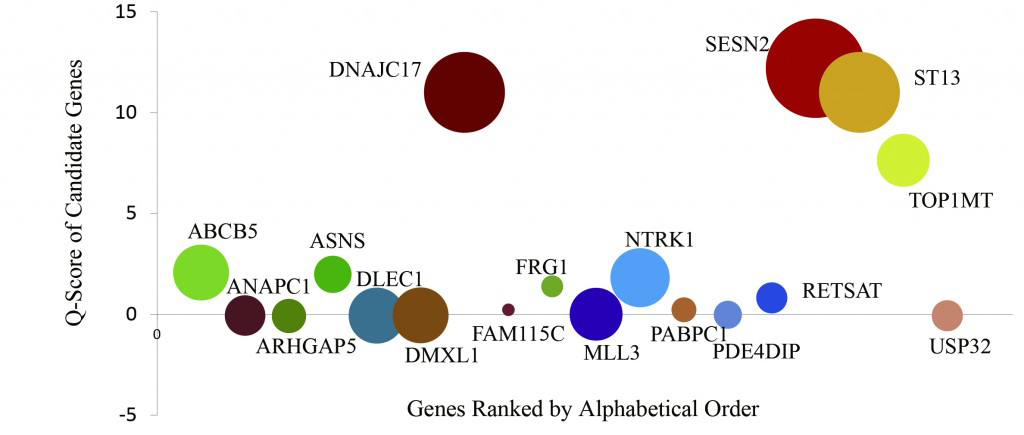
Figure 3. Key Gene Identification of the ET Patient
The driver gene prediction analysis of the 18 ET candidate genes is indicated as Q score. The vertical axis is the Q score, and the circle size (diameter) indicates the cell mutation frequency.
Other Publications:
- Xun X, et al. Single-Cell Exome Sequencing Reveals Single-Nucleotide Mutation Characteristics of a Kidney Tumor. Cell 2012,148:886-95.
- Zhang C, et al. A Single Cell Level Based Method for Copy Number Variation Analysis by Low Coverage Massively Parallel Sequencing. PLoS One 2013, 8(1):e54236.
- Yin X, et al. Massively Parallel Sequencing for Chromosomal Abnormality Testing in Trophectoderm Cells of Human Blastocysts. Biology of Reproduction 2013 Mar 21, 88(3):69.
Single cells are isolated from tissues. Every qualified cell is then lysed and genomic DNA is amplified by MDA, which has an error rate is 10-6-10-7. The following figure shows a detailed workflow for our single cell sequencing service, including whole genome re-sequencing, whole exome sequencing, and target region re-sequencing. Furthermore, BGI can also provide customized workflows to meet specific analysis needs.
Figure 4. Workflow of Single Cell Sequencing
Bioinformatics:
Standard Bioinformatics Analysis
- Data filtering (removing adaptors, contamination, and low-quality reads from raw reads)
- Alignment and summary of data production
- SNP calling, annotation, and statistics
- InDel calling, annotation, and statistics
- CNV calling, annotation, and statistics (only for whole genome resequencing)
- SV calling, annotation, and statistics (only for whole genome resequencing)
Custom Bioinformatics Analysis
We can also perform customized analysis to meet requirements of specific projects, e.g., sub-clone evolution of tumor.Sample Requirements:
- Fresh Tissue: A size of 1-2 cm3 is recommended; as low as 5 mm3 is also acceptable for precious samples. Tissue samples should be immediately stored in liquid nitrogen or at -80 ℃ after surgical resection, without other solvents treatment.
- Whole Blood (or Bone Marrow): The total volume should be no less than 5 ml. Samples should be collected with anticoagulant tube and stored at -80℃.
- Cell Suspensions: No fewer than 100,000 cells are recommended. It is necessary to follow the standard cell cryopreservation operation protocols, freeze cells gradually with cryopreservation media, and store them in liquid nitrogen or at -80℃.
- Isolated Single Cells: Single cells should be stored separately in 3-5 μL solvent (e.g., PBS), in DNase/Rnase free PCR tube (200 μL), and stored at -80 ℃ for no longer than one week. Cells should be free of nucleic acid–binding dyes.