
- Genomics
- Transcriptomics
- Epigenomics
- Meta-omics
- Proteomics
- Single-Cell Sequencing
- Immune Repertoire Sequencing
- FFPE Samples

Target Region Sequencing
Cases
Technical Information
Contact Us / Wish List
Target region capture enriches specific regions (e.g., the MHC region) or specific genes by probe hybridization based on probes designed according to the genomic regions of interest. It is cost effective to use targeted region sequencing to find variants with large samples. BGI has completed over 65,000 analyses using target region sequencing and has developed a series of software (e.g., SOAPsnp) to analyze the sequencing data and generate precise alignment results, accurate variance results, and custom analysis results. Many successful cases have been published in journals such as Science and Mammalian Genome.
Benefits:
- Targeted: Focus on the regions of interest, such as exons, promoters, and enhancers.
- Cost effective: Much lower cost for narrowed region sequencing, which is a significant advantage for projects with large sample sizes and deep sequencing.
- Wide applications: QTL fine mapping, association studies, result validation for large amount of samples, and clinical sequencing.
- High quality data (See examples of data generated by BGI).
- Rich experience: Our technicians have performed a large number of targeted region sequencing projects; they are familiar with experimental methods and trouble-shooting techniques.
- Custom-tailored: Custom bioinformatics analysis is available for specific research purposes and data characteristics. Internal software evaluation is available to update the analysis pipeline and ensure an optimized final analysis result for the customer.
Customer Testimonial:
"We use BGI as a NGS service provider for many of our targeted region re-sequencing projects. BGI has consistently provided high-quality data with a very quick turnaround time. I know exactly what to expect in terms of delivery time and data quality, which is vital for our business operations." -Dr. Eric Lin, Chairman of the Board, Otogenetics CorporationTargeted Next-generation Sequencing as a Comprehensive Test for Patients With and Female Carriers of DMD/BMD: a Multi-population Diagnostic Study. European Journal of Human Genetics. Doi: 10.1038/ejhg. (2013).
Duchenne and Becker muscular dystrophies (DMD/BMD) are the most commonly inherited neuromuscular diseases. However, accurate and convenient molecular diagnosis cannot be achieved easily because of the enormous size of the dystrophin gene and complex causative mutation spectrum. Here, we introduce a new single-step method for the genetic analysis of DMD patients and female carriers in real clinical settings and demonstrate the validation of its accuracy. A total of 89 patients, 18 female carriers and 245 non-DMD patients were evaluated using our targeted NGS approaches. We detected novel partial deletions of exons in nine samples for which the breakpoints were located within exonic regions. The results proved that our new method is suitable for routine clinical practice, with a shorter turnaround time, higher accuracy, and better insight into comprehensive genetic information (detailed breakpoints) for ensuing gene therapy.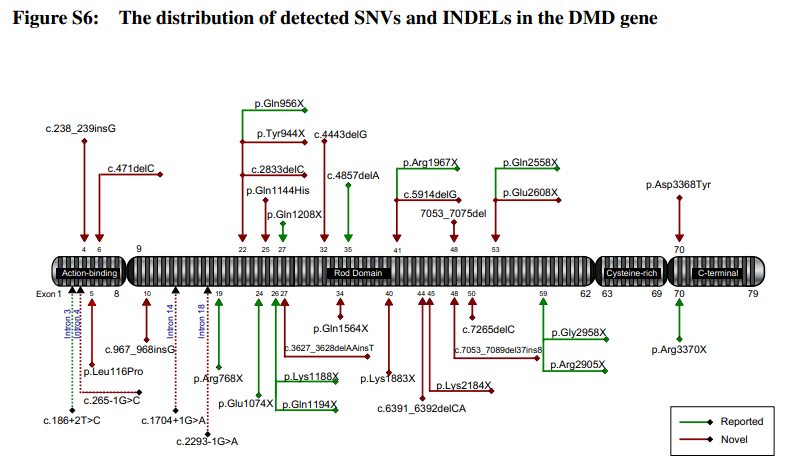{kind=link}
An Abundance of Rare Functional Variants in 202 Drug Target Genes Sequenced in 14,002 People. Science. 337(6090): 100-4. (2012).
Rare genetic variants contribute to complex disease risk. However, the abundance of rare variants in human populations remains unknown. This paper explored this spectrum of variation by sequencing 202 genes encoding drug targets in 14,002 individuals. The research found that rare variants are abundant (1 every 17 bases) and geographically localized, so that even with large sample sizes, rare variant catalogs will be largely incomplete. The paper used the observed patterns of variation to estimate population growth parameters, the proportion of variants in a given frequency class that are putatively deleterious, and mutation rates for each gene. The paper concludes that because of rapid population growth and weak purifying selection, human populations harbor an abundance of rare variants, many of which are deleterious and have relevance to understanding disease risk.{kind=link}
Bioinformatics:
Standard Bioinformatics Analysis
- Data filtering (removing adaptors contamination and low-quality reads from raw reads)
- Align reads to the human reference genome (UCSC build HG19) using BWA software
- Assessment of sequencing quality, including data production statistics, sequencing depth distribution and coverage uniformity
- SNP calling (tools: SAMtools, SOAPsnp, or GATK)
- SNP annotation (annotate each SNP to the corresponding gene functional units in RefGene database, including nucleotide and amino acid changes)
- SNP validation and comparison [with dbSNP database, 1000 Genomes Project database, publicly available exome databases (ESP), and YH (YH is only applied in Pan Asia-pacific region)]
- Functionality and conservation prediction of SNPs (based on SIFT, Polyphen-2, PhyloP, GERP score, Mutation assessor, Condel and FATHMM)
- Statistics of SNPs in each functional element
- InDel calling (tools: SAMtools or GATK)
- InDel annotation (annotate each InDel to the corresponding gene functional units in RefGene database, including nucleotide and amino acid changes)
- InDel validation and comparison [with dbSNP database, 1000 Genomes Project database, publicly available exome databases (ESP), and YH (YH is only applied in Pan Asia-pacific region)]
- Statistics of InDels in each functional element
Advanced Bioinformatics Analysis
General Advanced Analysis
- Non-coding SNP calling, annotation, and statistics
- Non-coding InDel calling, annotation, and statistics
Cancer Advanced Analysis
- Preliminary identification for the paired tumor-normal samples based on MassARRAY (recommended prior to sequencing)
- Somatic SNV and InDel calling, annotation, and statistics [somatic SNV and InDel will be compared against the public databases such as dbSNP, 1000 Genomes Project, and YH (YH is only applied in Pan Asia-pacific region)]
- Somatic SNV/InDel annotation against the COSMIC database
- Functionality and conservation prediction of somatic SNVs (based on SIFT, Polyphen-2, PhyloP, GERP score, Mutation assessor, Condel, and FATHMM)
- Non-synonymous annotation for mutated genes against the CancerGeneCensus database
Complex Diseases Advanced Analysis
- Sample design and power calculation (during project design stage)
- Population-level SNP calling and linkage disequilibrium (LD)-based genotype calling
- SNP annotation and statistics (including OMIM and ENCODE annotation)
- Quality control (QC) for population SNPs, including base quality, reads map quality, allele balance, DNA strand bias, homo-polymer, HWE test, and SNP filtering within 5bp around an InDel
- Sample QC, including cryptic relatedness analysis, sample contamination detection based on inbreeding coefficient, and PCA population stratification detection
- Single site SNP association test
- eQTL analysis
Population Advanced Analysis
- Population-level SNP calling, annotation, and statistics
- Quality control (QC) for population SNPs, including base quality, reads map quality, allele balance, DNA strand bias, homo-polymer, HWE test, and SNP filtering within 5bp around an InDel
- Genotype imputation and haplotype phasing analyses based on reference panels
- Sample QC, including relatedness detection and contamination detection based on inbreeding coefficient
- Selection signal detection, especially recent selection events, based on iHS and XP-EHH tests with validation using DDAF, Fst, and Tajima's D methods
- GO functional analysis, KEGG and panther pathway enrichment analyses for the candidate selected genes
Mendelian disorders Advanced Analysis
Please contact technical support([email protected]) for details.
Custom Bioinformatics Analysis
Customized Analysis of Complex Disease
(Applied to large-scale sample design, ≥200 cases & controls are recommended)- Gene-based association analysis (CMC, SKAT-O, WSS, KBAC, Collapse)
- Candidate gene prioritization
- Conditional analysis for candidate SNPs
- LD and haplotype block analyses involving candidate SNPs
- GO functional analysis and KEGG pathway enrichment analysis for the candidate selected genes
- Pathway-based protein interaction analysis (pairwise or multiple interaction)
- ROC curve plot and heritability estimation
- eQTL analysis
- Population-level CNV calling, annotation, and statistics
- Association analysis of CNVs (Merge & Split)
De novo mutation analysis based on family samples
- De novo SNV calling, annotation, and statistics
- De novo InDel calling, annotation, and statistics
- Association analysis of de novo SNVs/InDels with disease phenotypes
- GO functional analysis, KEGG pathway enrichment analysis, and protein-protein interaction analysis
- Parents tracing for de novo SNV mutations
(The following items are applied to multiple families, ≥30 trios or 10-20 quads are recommended)
Kits Available for Target Region Sequencing
Sample Requirements:
Human Target Region Sequencing
For the genomic DNA samples:- Purity:OD260/280= 1.8-2.0, without degradation and RNA contamination
- Concentration: ≥ 37.5ng/μl
- Quantity: 1μg (2.5 μg gDNA recommended)
Mouse Target Region Sequencing
For the genomic DNA samples:- Purity: OD260/280= 1.8-2.0, without degradation and RNA contamination
- Concentration: ≥ 37.5 ng/μl
- Quantity: 1μg (2.5 μg gDNA recommended)