

Comparative Analysis of the Proteome and the Transcriptome
Cases
Technical Information
Contact Us / Wish List
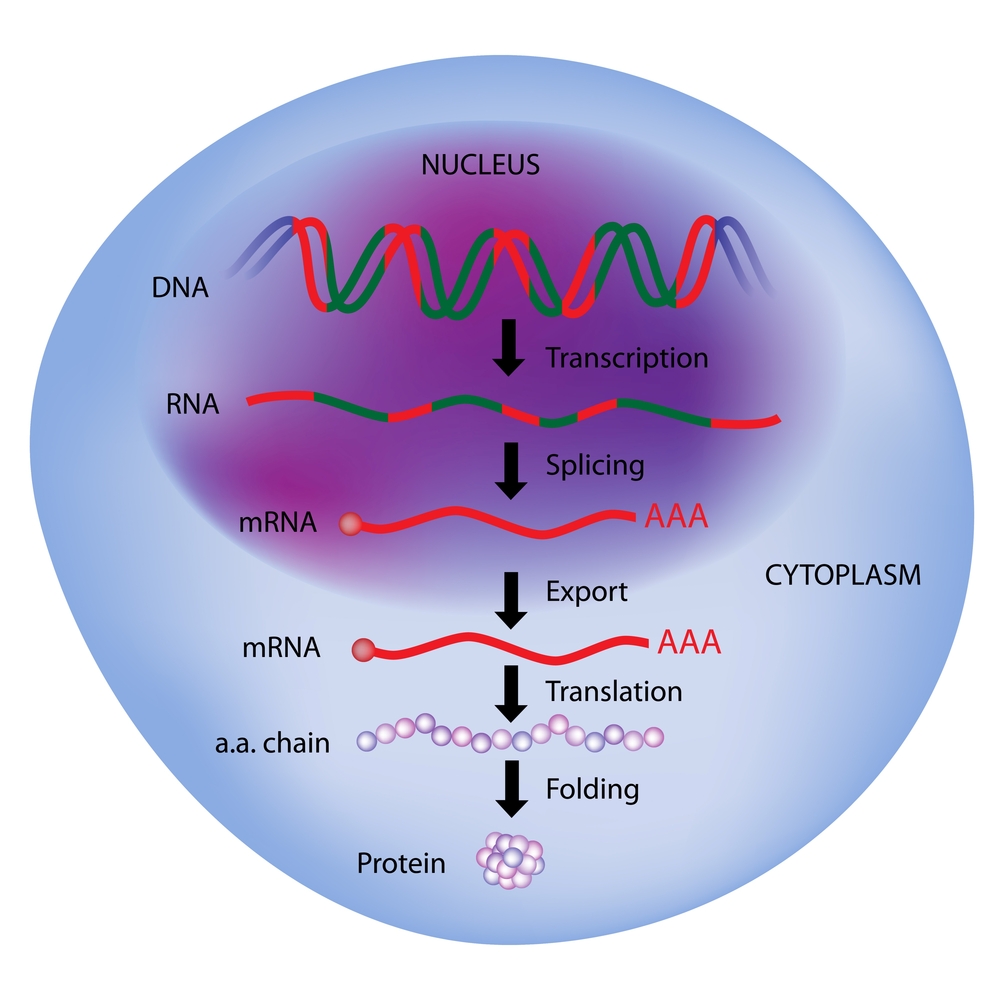 Protein is the carrier and function executor of life activities. The different expression levels of proteins play an important role in cell growth, environment stimulation reaction and disease development as well. Moreover, there is a delicate gene expression mechanism governing the process from DNA, mRNA to protein, such as transcriptional regulation, post-transcriptional regulation, translational regulation and post-translational regulation. Therefore, monitoring the expression level of mRNA and protein synchronously (or simultaneously) and performing integrated analysis have become an inevitable trend for studying molecular basis of diseases, exploring the mechanism of environmental response and discerning key gene expression patterns.
Protein is the carrier and function executor of life activities. The different expression levels of proteins play an important role in cell growth, environment stimulation reaction and disease development as well. Moreover, there is a delicate gene expression mechanism governing the process from DNA, mRNA to protein, such as transcriptional regulation, post-transcriptional regulation, translational regulation and post-translational regulation. Therefore, monitoring the expression level of mRNA and protein synchronously (or simultaneously) and performing integrated analysis have become an inevitable trend for studying molecular basis of diseases, exploring the mechanism of environmental response and discerning key gene expression patterns.
Benefits:
- Obtaining a comprehensive understanding of gene expression (e.g. key gene expression patterns)
- Unveiling novel gene regulatory mechanisms through analyzing expression level of mRNA and corresponding proteins
- Providing valuable inputs in protein identification from transcriptome database for some species lacking complete protein database
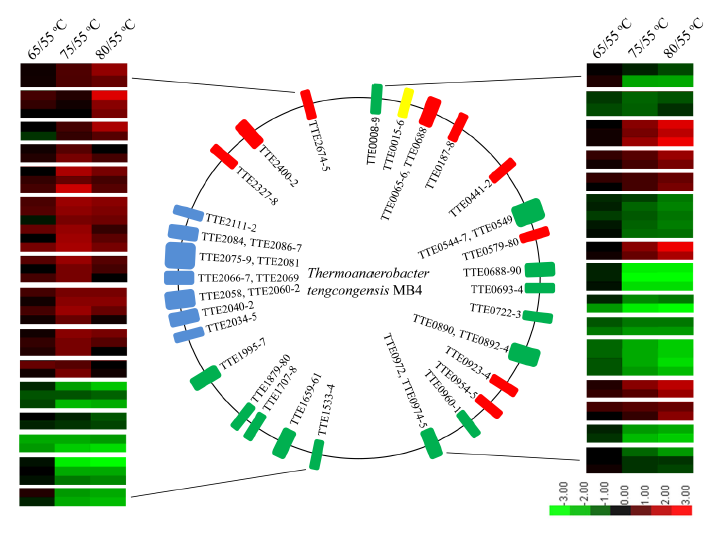
Quantitative Proteomics Reveals the Temperature-dependent Proteins Encoded by a Series of Cluster Genes in Thermoanaerobacter tengcongensiss. Mol Cell Proteomics. 12: 2266-2277 (2013).
{kind=link}
Comprehensive and quantitative information of the thermophile proteome is an important source for understanding of the survival mechanism under high growth temperature. Thermoanaerobacter tengcongensis (T. tengcongensis), a typical anaerobic thermophilic eubacterium, was selected to quantitatively evaluate its protein abundance changes in response to four different temperatures. With optimized procedures of isobaric tags for relative and absolute quantitation quantitative proteomics (iTRAQ), such as peptide fractionation with high-pH reverse phase (RP) high performance liquid chromatography (HPLC), tandem MS acquisition mode in LTQ Orbitrap Velos MS, and evaluation of the quantification algorithms, high quality of the quantitative information of the peptides identified were acquired. In total, 1589 unique proteins were identified and defined 251 as the temperature-dependent proteins. Analysis of genomic locations toward the correspondent genes of these temperature-dependent proteins revealed that more than 30% were contiguous units with relevant biological functions, which are likely to form the operon structures in T. tengcongensis. The RNA sequencing (RNA-seq) data further demonstrated that these cluster genes were cotranscribed, and their mRNA abundance changes responding to temperature exhibited the similar trends as the proteomic results, suggesting that the temperature-dependent proteins are highly associated with the correspondent transcription status. Hence, the operon regulation is likely an energy-efficient mode for T. tengcongensis survival.
Data Applicable to Integrated Analysis:
- Proteomics data from iTRAQ, label-free quantification, and 2D gel band and spot identification
- Transcriptomics data from RNA-Seq (de novo), transcriptome resequencing, digital gene expression (DGE), and microarray
Bioinformatics:
- The qualitative relationship between the proteome and the transcriptome
- Expression correlation between the proteome and the transcriptome
- Expression pattern cluster analysis of multiple samples between the proteome and the transcriptome