

RNA-Seq (Quantification)
Cases
Technical Information
Contact Us / Wish List
 RNA-Seq (Quantification) is used to analyze gene expression of certain biological objects under specific conditions. It is a cost-effective quantification method that produces transcripts quantification results of greater sensitivity, higher reproducibility, and wider dynamic range. BGI quantifies genome-wide gene expressions using next-gen sequencers Illumina HiSeq and Ion Proton, and provides differential gene expression and pathway analysis. Powered by our unique bioinformatics capabilities, BGI’s RNA-Seq services deliver reliable data and analyses to our customers conducting drug response studies, biomarker detection, basic medical research, and drug R&D.
RNA-Seq (Quantification) is used to analyze gene expression of certain biological objects under specific conditions. It is a cost-effective quantification method that produces transcripts quantification results of greater sensitivity, higher reproducibility, and wider dynamic range. BGI quantifies genome-wide gene expressions using next-gen sequencers Illumina HiSeq and Ion Proton, and provides differential gene expression and pathway analysis. Powered by our unique bioinformatics capabilities, BGI’s RNA-Seq services deliver reliable data and analyses to our customers conducting drug response studies, biomarker detection, basic medical research, and drug R&D.
Check out our hot rapid RNA-Seq (Quantification) offer on the Ion Proton platform, taking you from sample to answer in only 9 business days for up to 40 samples.
Benefits:
- Strong bioinformatics analysis capabilities on RNA-Seq (Quantification) data
- High reproducibility: Over 0.99 of correlation coefficient between sample replicates.
- Two platforms, Illumina Hiseq and Ion Proton, are available depending on the specific needs.
- RNA-Seq (Quantification) based on Hiseq: high-throughput and a wide range of read lengths (50 ~150 bp read lengths are available)
- RNA-Seq (Quantification) based on Ion Proton: rapid turnaround time (only 9 business days for up to 40 samples) and longer read sequencing length (average read length is 150 bp).
- Wide detection range: Accurate quantification of expression levels of over 6 orders of magnitude of transcript abundance
- High accuracy: High concordance between RNA-Seq (Quantification) and qPCR or microarray results.
Customer Testimonials:
"As the principal investigator on two sequencing projects that are pushing the state-of-the-art (viral discovery using metagenomic sequencing and phylogenomic analysis of 1000 plant species), I cannot expect everything to work on the first iteration but I do expect my sequencing provider to work together with me to correct whatever went wrong regardless of whose fault it was. BGI-Shenzhen does that admirably."
- Dr. GaneKa-Shu Wong, Professor and iCORE Chair in Biosystems Informatics Department of Biological Sciences, University of Alberta
BGI has validated the reproducibility, sensitivity, and detection range of RNA-Seq (Quantification) on both platforms.
Some results from Illumina platform are as follows. (Please click here for the validation results from the Ion Proton platform):
Materials: Human Brain Reference RNA (HBRR) from FirstChoice ®;
Universal Human Reference RNA (UHRR) from Stratagene
Standardized method of gene expression analysis: RPKM (Reads per kb per million reads) is used to quantify the gene expression level. The method is able to eliminate the influence of different gene lengths and sequencing discrepancy in the measurement.
High Reproducibility

Figure 1: Technical Replicate Results of RNA-Seq (Quantification). From the technical replicate data, the spearman R is 0.992 and 0.993, respectively.
The results suggest that RNA-Seq has high reproducibility.
High Accuracy

Figure 2: Correlation between results from RNA-Seq (Quantification) and qPCR RNA-Seq is a highly accurate quantitation method. The scatter-plot of figure 2 shows a high correlation between results from RNA-Seq and qPCR. Spearman R is 0.915 and slope is 1.022.
High Sensitivity

Figure 3: Detection Range of RNA-Seq (Quantification) is wider than microarray, and RNA-Seq can detect more low abundance genes than microarray.
Analysis of Transcriptome Differences between Resistant and Susceptible Strains of the Citrus Red Mite Panonychus citri (Acari: Tetranychidae). Plos One. DOI: 10.1371. (2011).
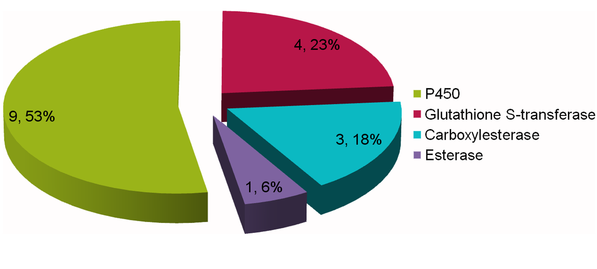 The citrus red mite is a worldwide citrus pest and a common sensitizing allergen of asthma and rhinitis. It has developed strong resistance to many registered acaricides, However, the molecular mechanisms of resistance remain unknown. In this study, a comparative transcriptome study was carried out in the susceptible strain (SS) and resistant strain of the Citrus red mite to study mite resistance. 2,701 differentially expressed genes (DEGs) based on the uniquely mapped reads were identified by the comparing the differences between RS and SS. Moreover, we identified 211 metabolism genes and target genes related to general insecticide resistance such as P450 and Cytochrome b, and further compared their differences between RS and SS.
The citrus red mite is a worldwide citrus pest and a common sensitizing allergen of asthma and rhinitis. It has developed strong resistance to many registered acaricides, However, the molecular mechanisms of resistance remain unknown. In this study, a comparative transcriptome study was carried out in the susceptible strain (SS) and resistant strain of the Citrus red mite to study mite resistance. 2,701 differentially expressed genes (DEGs) based on the uniquely mapped reads were identified by the comparing the differences between RS and SS. Moreover, we identified 211 metabolism genes and target genes related to general insecticide resistance such as P450 and Cytochrome b, and further compared their differences between RS and SS.
Bioinformatics:
Standard Bioinformatics Analysis
- Data filtering includes removing adaptors, contamination and low-quality reads from raw reads
- Assessment of sequencing
- Gene expression annotation
- Condition specific expression analysis (Only for the Proton platform)
- Principle component analysis (Only for the Proton platform)
- Differential gene expression analysis
- Expression pattern analysis of differentially expressed genes (DEGs)
- Gene ontology enrichment analysis of DEGs
- Gene ontology classification (WEGO analysis)
- Pathway enrichment analysis of DEGs
- Protein-protein interaction network analysis
Custom Bioinformatics Analysis
- We can also perform customized analyses to meet the requirements of specific projects.
Sample Requirements:
- Sample condition: Integrated total RNA samples that have been treated with DNase; Avoid protein contamination during RNA isolation
- Sample quantity (for library construction):
- Illumina platform: Total RNA ≥ 2 µg (human, rat and mice), Total RNA ≥ 5 µg (others)
- Ion Proton platform: Total RNA ≥ 1 µg (human, rat and mice), Total RNA ≥ 5 µg (others)
- Sample concentration:
- Illumina platform: ≥ 80 ng/µL (human, rat and mice), ≥ 200 ng/µL (others)
- Ion Proton platform: ≥ 80 ng/µL (human, rat and mice), ≥ 200 ng/µL (others)
- Sample purity: OD260/280 = 1.8-2.2; OD260/230 ≥ 1.8; for animal RIN ≥ 7.0, for plant and fungi RIN ≥ 6.5; 28S:18S ≥ 1.0
Turnaround Time:
Ion Proton platform: Only 9 business days after samples pass QC for up to 40 samples
Illumina platform: ~30 business days after samples pass QC for up to 20 samples